
Proteínas e Inteligencia Artificial: ¿Por qué Google dice que ha revolucionado la biología moderna?
Cuando un programa de aprendizaje profundo, también llamada Inteligencia Artificial, cumple sus objetivos, los medios se apresuran a difundir la noticia como si las máquinas estuvieran ganando una guerra contra el ser humano. Nada más falso que esto, la IA es tan solo una herramienta más de programación, diseñada por humanos para resolver problemas complejos.
Tampoco debe sorprender que sobre todo se mediaticen los resultados de equipos de investigación que están asociados a las grandes empresas tecnológicas. Por eso es que cuando una tecnología desarrollada por Google, Microsoft, Apple o alguna otra multinacional obtiene buenos resultados se vuelven de dominio público algunos temas científicos que antes eran abordados por unos pocos especialistas.
Este fenómeno de visibilizar áreas de investigación muy reducidas puede resultar bueno, pero también debemos ser cuidadosos con el optimismo desmedido con el que por lo general se difunden estas notas. No hay que perder de vista que al final de todo, la publicidad para estas tecnologías se transforma en inversiones millonarias que pueden llevar a sostener agendas increíblemente absurdas como los viajes turísticos espaciales que hoy están en boca de todos, gracias a lo que algunos han llamado “una guerra de meadas entre machos multimillonarios”.
Más real y auténticamente aplicable es lo que acaba de lograr el programa de aprendizaje automático AlphaFold, de la compañía de Google DeepMind, que según sus creadores “acaba de sacudir los cimientos de amplios sectores de la ciencia al dar soluciones al problema mucho mejores de lo logrado en cinco décadas de intentos.”
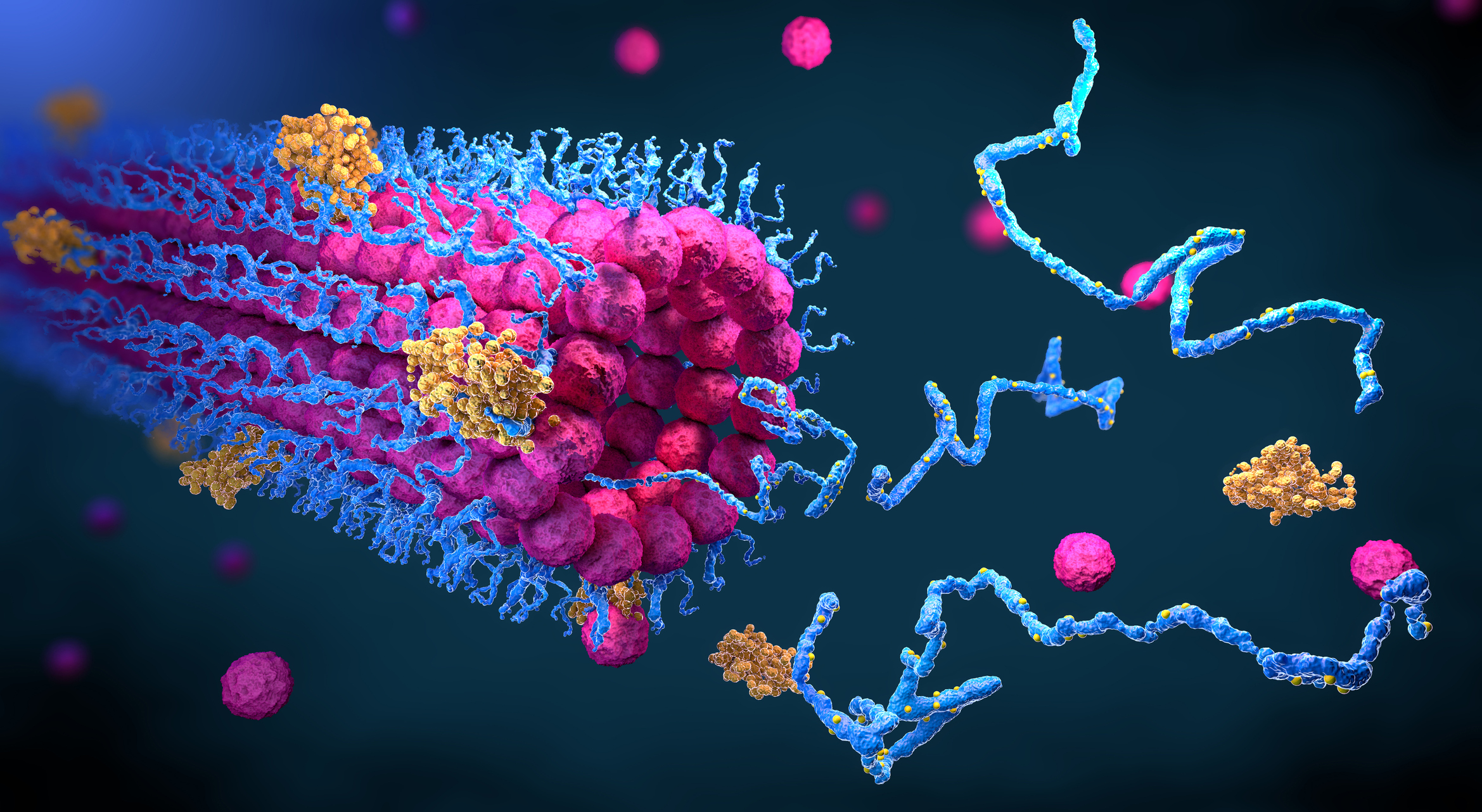
Si hay un problema capaz de enamorar a biólogos, físicos, matemáticos, informáticos, químicos y hasta médicos, es el de cómo se pliegan las proteínas: cómo adquieren su estructura tridimensional, su forma. Resolver el reto ayudará a entender un sinfín de procesos biológicos y acelerará la obtención de nuevos fármacos, entre otros codiciados beneficios.
Conocer la estructura de las proteínas, su forma, es importante. Las proteínas son las nanomáquinas del organismo y trabajan encajando unas en otras, como en un microscópico Tetris tridimensional. Si se sabe la forma de las proteínas es más fácil entender su función, y también cómo bloquearlas con fármacos en caso necesario.
Pero determinar la estructura de las proteínas es muy trabajoso; habitualmente implica cristalizarlas y analizar con rayos X la disposición de sus átomos. En cambio hoy es relativamente sencillo hallar la secuencia lineal de las piezas químicas que componen la proteína, los aminoácidos, y por eso el sueño de los biólogos es aprender a traducir la secuencia lineal en estructura tridimensional.
Viene a ser como predecir la forma final de una joya conociendo solo las gemas que la componen y el orden en que van colocadas.
“La información para el plegamiento está en la secuencia, eso lo sabemos; una proteína que ha perdido su estructura, en las condiciones adecuadas vuelve a plegarse bien”, explica Alfonso Valencia, director de Ciencias de la Vida del Centro de Supercomputación de Barcelona, interesado en este problema casi desde el inicio de su carrera. “¿Qué leyes físicas rigen el proceso? Lo que queremos es entenderlas porque así tendríamos una fórmula, la aplicaríamos, y ya; pero no lo hemos conseguido”.
En los años 80 y 90 los ordenadores entraron a escena, logrando avances modestos. Para estimular el área, en 1994 el biólogo computacional John Moult, de la Universidad de Maryland (EE UU), creó CASP (siglas en inglés de Evaluación Crítica de Predicción de Estructuras), un concurso bienal en el que grupos de todo el mundo ponen a prueba sus programas de predicción de estructuras. El reto consiste en que, en cada edición, cristalógrafos anuncian, pero no publican, las estructuras que van resolviendo, y mientras tanto los bioinformáticos ponen a trabajar sus programas para tratar de predecirlas. Ganan, obviamente, las predicciones más próximas al resultado experimental.
AlphaFold ya participó en 2018 con muy buenos resultados. Pero nada comparado con el éxito abrumador de esta edición, que acaba de concluir. “Estamos todos asombrados, la diferencia con los demás grupos ha sido abismal, estratosférica”, dice Valencia, que este año formaba parte de la quincena de evaluadores de CASP. Matiza que más que contento está “interesado”, y que “aún lo estoy procesando”.
Moult ha declarado a la revista Nature que, “en cierto sentido, el problema [del plegamiento de proteínas] está resuelto”. Nature cita también a Janet Thornton, bióloga estructural del Laboratorio Europeo de Biología Molecular (EMBL) y asesora en CASP, que añade: “Empezaba a creer que no vería resuelto este problema en mi vida”.
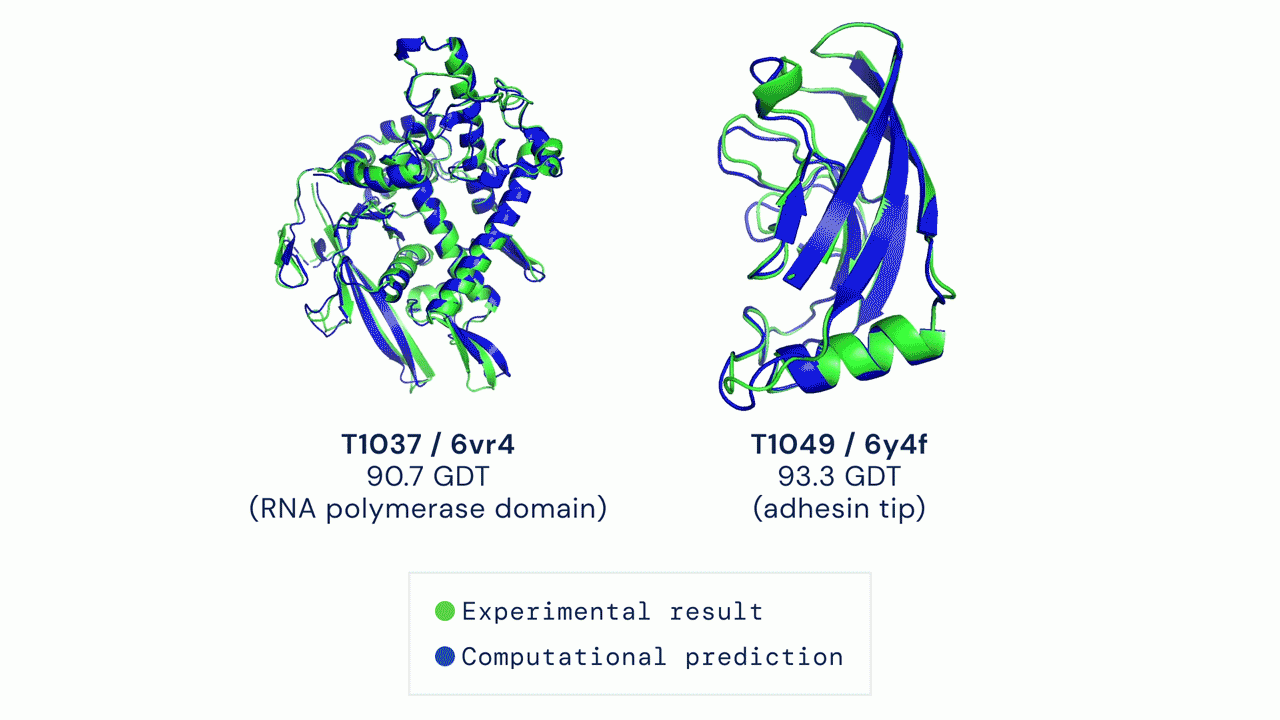
Predicciones indistinguibles de la realidadAlphaFold competía en la categoría más difícil, la de proteínas sobre cuya estructura no hay ninguna pista. Junto a otros 60 grupos de todo el mundo el programa hizo predicciones para más de un centenar de proteínas. Algunas resultaron casi indistinguibles de las estructuras consideradas reales, y solo en dos casos se alejaron bastante.
Ahora el equipo de AlphaFold, de unos treinta investigadores, prepara una publicación con detalles técnicos que tardará meses en ver la luz. Mientras tanto han explicado su trabajo en una ponencia, y en el resumen que, como todos los participantes, deben presentar a CASP.
En un trabajo divulgativo publicado el mismo día de la resolución de CASP afirmaban: “Nuestras predicciones tienen un error medio de aproximadamente 1,6 angstroms, lo que es comparable al grosor de un átomo. (…) Estos emocionantes resultados abren para los biólogos la posibilidad de usar la predicción computacional de estructuras como una herramienta clave en investigación científica. Nuestros métodos pueden ser especialmente útiles para clases importantes de proteínas, como las de membrana, que son muy difíciles de cristalizar y cuya estructura es por tanto compleja de determinar experimentalmente”.
¿Cómo lo han conseguido? La inteligencia artificial en sí no es lo que ha marcado la diferencia, dado que todos los grupos recurren ya a técnicas de aprendizaje automático. Se diría que tampoco ha sido clave el pertenecer a una gran compañía: Microsoft y la china Tencent también competían. Para Valencia, los de AlphaFold “simplemente son muy buenos, no es que tengan algo nuevo muy secreto”.
Redes neuronales entrenadas con 170.000 proteínasA falta de más detalles, este equipo ha explicado que han recurrido a un tipo de redes neuronales distintas a las empleadas en 2018, un sistema —al que llaman AlphaFold2— que además del aprendizaje automático incorpora información adicional sobre la física y la geometría del proceso de plegamiento. Su abordaje tiene aspectos novedosos, como que en lugar de predecir las relaciones entre aminoácidos, las redes neuronales predicen directamente la estructura final. Es un sistema más complejo que “produce estructuras de proteínas mucho más precisas”, indican los autores en el libro de resúmenes de CASP.
AlphaFold2 aprendió entrenándose durante semanas con 170.000 proteínas cuya estructura se conoce y está disponible en bases de datos públicas. Predecir cada estructura le lleva unos días. No necesita una capacidad de computación superior a la habitual en estos programas.
¿Se han quedado sin trabajo los cristalógrafos? No parece probable al menos a corto plazo, porque seguirá siendo indispensable confirmar las estructuras. Pero AlphaFold ahorrará seguramente años de trabajo, poniendo sobre la pista a los experimentales. Para Valencia, “se tardará aún un poco en ver las implicaciones prácticas, pero llegarán y tendrán mucho impacto”.
Thorton cree, para empezar, que el nuevo programa ayudará a desvelar la función de miles de proteínas humanas cuya estructura aún se desconoce, así como a entender por qué bastan pequeñas variaciones en algunas proteínas para causar enfermedades.
El problema de fondo sigue Demis Hassabis, fundador de DeepMind, ha tuiteado que este éxito “representa para nosotros una emocionante primera demostración” de que la inteligencia artificial desarrollada en DeepMind sirve para “acelerar el descubrimiento científico”, algo que “siempre ha sido la visión última” de esta compañía. Los logros previos de DeepMind han sido en otros ámbitos, como cuando uno de sus programas batió al ganador mundial del complejo juego Go.
Los biólogos estructurales se preguntan, no obstante, si la compañía publicará el código de AlphaFold2. De la primera versión de AlphaFold sí se publicó información suficiente como para que los demás grupos pudieran replicar la estrategia. Es algo que muchos consideran adecuado puesto que, al fin y al cabo, estos programas aprenden gracias a las secuencias y estructuras halladas por toda la comunidad a lo largo de décadas.
Aun reconociendo sus logros, hay algo que no todos están dispuestos a conceder a AlphaFold: los laureles de haber resuelto un problema estrella de la biología. David de Sancho, experto en estructura de proteínas en la Universidad del País Vasco, opina en Twitter que el programa ha resuelto “la predicción de estructuras”, “no el plegamiento de proteínas tal como lo entiende la mayoría” de quienes trabajan en el área. Esto último implicaría “predecir los pasos que sigue la proteína para pasar del estado desplegado al plegado”, algo que nadie ha conseguido.
Escucha nuestro podcast
También Valencia cree que AlphaFold proporciona un atajo muy valioso en la práctica, pero no resuelve el problema de fondo: “Predecir estructuras no es equivalente a resolver el problema científico de cómo se pliegan las proteínas”.









{kind=link}